


 Автор : к.мед.н. невропатолог Олексюк-Нехамес Алла Григорівна
Автор : к.мед.н. невропатолог Олексюк-Нехамес Алла Григорівна
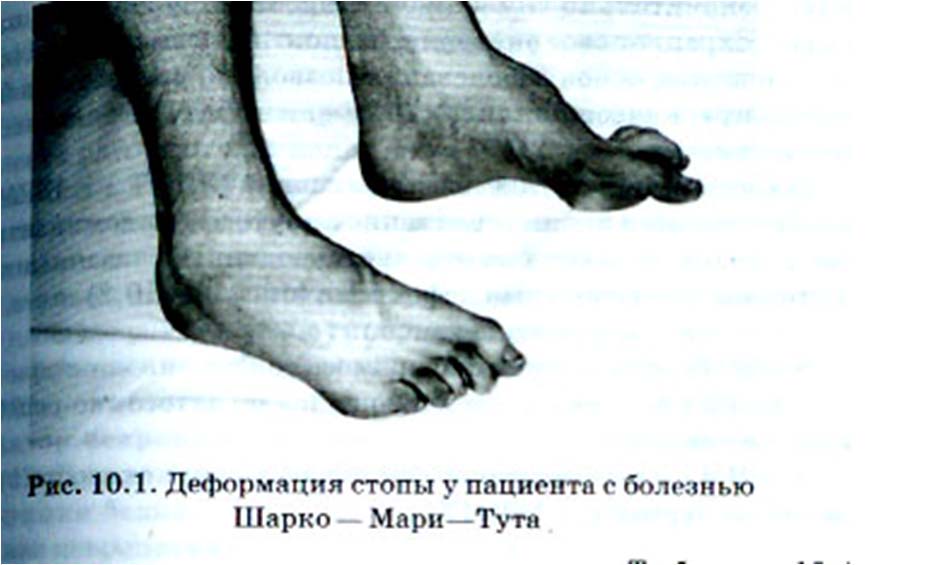
Спадкові моторні і сенсорні невропатії.Елекрофізіологічні дослідження і біопсія нервів дозволяє диференціювати декілька варіантів спадкових моторних та сенсорних невропатій як з домінантним і з рецесивним типом успадкування.Виділяють 7 типів спадкових моторних та сенсорних поліневропатій.Класифікація спадкових моторно-сенсорних невропатій. Порівняльна характеристика основних типів спадкових моторно-сенсорних невропатій. Класифікація спадкових поліневропатій. Виділяють наступні форми спадкових невропатій: спадкові моторно-сенсорні невропатії 2.Спадкові сенсорно-вегетативні невропатії 3.Сімейні амілоїдні поліневропатії 4.Інші спадкові поліневропатії — (адреномієлоневропатії, хвороба Фабрі, танжерська хвороба, сімейна гігантська аксональна невропатія, невропатія повязана з множиними ендогенними пухлинами 2В типу) В МКХ -10 спадкові поліневропатії представлені в рубриці G 60 і Е85 (спадкові ідіопатичні невропатії). До групи спадкових моторних і сенсорних невропатій відносять: 1.Хвороба Шарко-Марі-Тута 2.Хвороба Дежеріна-Сотта 3.Спадкова моторна і сенсорна невропатія І- VII типу (невральна аміотрофія) 4.Гіпертрофічна невропатія у дітей 5.Перонеальна мязева атрофія(аксональний тип) гіпетрофічний тип і 6.синдром Руссі-Леві Хвороба Шарко-Марі-Тута – демієлінізуючий варіант з вираженим дистальним тетрапарезом і амітрофією , з повільним протіканням і прогресуваннням. Хвороба Ревсума або СНМС IV типу кодується в рубриці G.60.1. характеризується повільно розвинутою слабістю, атрофіями м’язів і зниженням чутливості в дистальних відділах кінцівок, арефлексією. Деформація стопи у пацієнта з хворобою Шарко-Марі-Тута. Хвороба Ревсума – аутосомно-рецесивне захворюваня повязане з порушенням метаболізму і накопиченням в організмі фітинової кислоти. Захворювання проявляється в дитячому віці або в юнацькому – у вигляді демієлінізуючої сенсо-моторної поліневропатії, прояви мозочкової атаксії, пігментної дегенерації сітківки, катарактою, іхтіозом (як звикло в області гомілок), аносмією, нейросенсорною тугоухістю. Протікання – повільне прогресуюче. Діагноз підтверджується вмістом фітанової кислоти в крові. Спадкова сенсорно-вегетативна невропатія 1-ого типу протікає з порушенням больової і температурної чутливості в нижніх кінцівках, тривало не загоєними трофічними виразкуваннями правої стопи, нейроартропатією суглобів обох стоп, має повільно протікаючий перебіг. СВС полінейропатія ІІІ типу – хвороба Райлі-ДЕЯ – в рубриці G.90.1. (до групи захворювання вегетативної нервової системи). Хвороба Фабрі відноситься до рубрики E75.2 – Анальфаліпопротеінемія – танжерська хвороба – до рубрики E78.6 – спадкові нейрометаболічні захворювання – відносять до ліпідемій, (див. наступний слайд. ) Гіперліпідемії виявляються у 10-20 % дітей та у 40-60 % дорослих. Вони можуть бути первинними, генетично детермінованими, або розвиваються вторино на фоні порушення дієти, різних захврювань, що ведуть до метаболічним розладів (інсулінзалежний діабет, хронічний панкреатит, алкоголізм, цироз печінки нефроз, дисглобулінемії та інші). Основні форми порушення обміну ліпопротеїдів: Сімейні ліпопротеінемії (генетичн детерміновані) абеталіпопротеінемії; гіпобеталіпопротеінемії; Анальфаліпопротеінемії (танжерська хвороба) Первинні гіперліпопротеінемії (I-V типи) Вторинні гиперліпопротеінемії Ліпідози: сфінгомієлінози ( хвороба Німанна-Піка); глюкоцереброзідоз ( хвороба Гоше); метахроматичні ліподистрофії (сульфатидліпідози); церемідтригексідоз ( хвороба Фабрі). Невроліпідози. ліпідозні невропатії. Невроліпідози – це спадкові хвороби ліпідозного обміну з переважним ураженням нервової системи. В метаболізмі нервової системи, особливо в процесах мієлінізації , серед складних ліпідів важливу роль грають похідні сфінгозина (сфінголіпіди). Метаболічні дефекти виявлені при декількох спадкових невропатіях: спадкова атактична поліневорпатія (хвороба Рефсума), метахроматична лейкодистрофія, γ— β — ліпопротеінемії,хвороба Фабрі.Танжерска хвороба — рідкісне порушення, що характеризується вираженною недостатністю або відсутністю повноцінних ЛПВП в плазмі і накопиченням ефірів холестерина у багатьох многих тканинах організму.Це захворювання отримало свою назву від острова Танжер в штаті Вірджинія (США), де вперше воно було виявлено. До сьогодня, в США, Европі і Австралії описані всього 26 випадків захворювання.Клінічні прояви.Підозра на танжерску хворобу частіше всього проявляється при незвичному вигляді миглдалин, які стають збільшеними , дольчастими, помаранчевого забарвлення, в результаті відкадання ефірів холестерина в їх тканинах. Відкладання цих ліпідів в інших тканинах обумовлює виникнення сплено- (8%) і гепатомегалію. (30 %), а також лімфаденопатію (20%), що може викликати підозру на новоутвір. Ефіри холестерина відкладаються в слизовій прямої кишки яка при ректороманоскопии виглядає як усіяна плямами помаранчевого коляру розміром 1-2 мм. При біопсії прямої кишки, показаної у всіх подозрілих випадках, в слизовій і підслизовому прошарку виявляють гістіоцити, переповнені піноподібними ефірами холестерину. У хворих у віці старше 40 років при дослідженні з допомогою щілинної лампи як звикло виявляють і інфільтрацію рогівки, яка, проте зір не порушує.У хворих з танжерскою хворобою зустрічаються різні неврологічні симптоми, які можуть служити основною причиною для звертання до лікаря. До цих симптомів часто відносяться слабість, парестезії, диплопія і надмірна пітливість. Обєктивно відмічається зниження мязової сили снижение мышечной силы с швидким , сповільненням глубоких сухожилкових рефлексів, окоруховимі парези, швидке виснаження і вибіркова втрата больової та температурної чутливості.Електроміографічне дослідження виявляє ознаки денервації уражних мязів.Нервова провідність при цьому зебережена. Хоча у двох хворих з танжерскою хворобою у віці 43 років спостерігали стенокардію, у інших ознаки ураження коронарних або периферичних сосудин відсутні були до віку 60 років. Питання в тому, чи відчувають хворі з танжерскою хворобою підвищений ризик розвитку передчасного атеросклерозу, заалишається відкритим. Якщо не ввжажати варіабельних по ступені неврологічних порушень, танжерска хвороба відноситться до доброякісним по протіканні.Хвороба Фабрі (Fabry, диффузна універсальна ангіокератома, спадковий дистонічний ліпідоз) — врожденне захворювання, що характеризується спадковим дефіцитом фермента альфа-G4-галактозідази, що приводить до накопичення гликоліпідів (цераміда) в цитоплазмі і лизосомах клітин різних органів і тканин.Описана вперше в 1898 г. англійським дерматологом Андерсоном (Anderson) и німецким дерматологом Фабрі (Fabry).Що провокує хворобу Фабрі: Хвороба Фабрі обусловлена недостатністю лізосомної гідролази — a-галактозідази А, успадковується по Х-счепленому рецессивному типу, в звязку з чим клінічна симптоматика проявляеться у осіб чоловічої статі. Ген a-галактозідази А локалізований на довгому плечі Х-хромосоми Xq22 ® q24.Патогенез Хвороби Фабрі:Ензимний дефект приводить до системного відкладання сфінгогліколіпідів і глікопротеїда в ураженних тканинах, в особливо в ендотелії і гладкій мускулатурі судин, сердці, нирках (нирокові клубочки і канальці), очах (епітеліальні клітини рогівки), гангліях вегетативної нервової системи.Симптоми Хвороби Фабрі:У гомозиготних мужчин захврювання проявляеться в дитячому віці або у підлітків так званими «кризами Фабрі», що характеризуються нестерпними болями пекучого характеру в долонях і стопах.Приступи болю можуть тривати декілька днів, і сопроводжуватися невеликою лихоманкою і підвищеннямШое. Іноді помилково діагностується ревматизм.Акропарастезії з віком стають більш частими і важкими.В дитячому віці відмічені характерні для хвороби Фабрі ураження шкіри – ангіокератоми (телеангіектазії або маленькі поверхневі ангіоми ), які локалізуються симметрично на стегнах, спині, сідницях, зовнішніх статевих органах, в області колінних суглобів і пупка.Ангіокератоми можуть бути плоскими або дещо піднятими над поверхнею шкіри, від темно-червоного до блакитно–чорного кольору, не стають блітнішими при натискуванні.Спадкові невропатії.(узагальнена класифікаційна картина, що пояснює особливості спадкових невропатій) СПНП — I, II типів- хвороба Шарко-Марі-Тута (найбільш частий варіант) I-типа (демієлінізуючий варіант): -проявляеться до 20 років — АД-типу (рідше АР) — повільно наростаюча слабість та симетрична слабість в дистальних відділах ніг, виражені атрофії — «ноги бусла» «пола стопа»)- атрофія рук — угнітання сухожилкових р-сів, легкі чутливі порушення , постуральний тремор в руках, потовщення нервовових стовбурів, II-типа (аксональний варіант) відрізняється суттєвим зниженням швидкості проведення по моторній порції нерва, потовщення стовбурів, рідко — «пола стопа»,IV типа (хвороба Рефсума), — АР типа — до 30 років.Повільно протікаюча поліневропатія, атаксія,- пігментна дегенерація сітківки,- глухота,- іхтіоз,- кардіоміопатія,- дієта з обмеженням фітина призупиняє захворювання. Спадкові сенсорно-вегетативні невропатії. частіше — I типа: — АД тип успадкування — початок на другому десятилітті — переважно порушення больової і температурної чутливості в нижніх кінцівках—> охоплення глибокої чутливості , випадіння сухожилкових р-сів, поява незагоєних трофічних виразок на стопах, мутиляція пальців. Спадкові поліневропатії. це гетерогенна група хворіб, успадкованих по: аутосомно-домінантному (АД), аутосомно-рецессивному і Х-счепленому з статтю типами. 1. спадкова сенсомоторна ПНП (хвороба Шарко—Мари—Тута). Виділяють два варіанта цього захворювання: 1)I тип (демієлінізуючий) зустрічаються у 66,2% хворих 2)II тип (аксональний) – в 23% всіх випадків. Клінічно обидва варіанти схожі і розрізняються результатами ЕНМГ. Захворювання починається в 10—16 років і характеризуеться тріадою симптомів: 1.атрофія кистей і стоп 2.розлади поверхневих видів чутливості 3.гіпо- або арефлексія . У хворих часто формуються деформації стоп (фрідрейхівські, полі, еквіноварусні). У деяких пацієнтів спостерігається помірно прогресуюче, висхідне протікання. Руки охоплюються пізніше і уражені в меньшій ступені. Характерний вираженний міжсімейний і внутрішньосімейний поліморфізм.2. Хвороба Дежеріна—Сотта (III тип) і хвороба Рефсума (IV тип) починається з перших років життя і проявляється руховими порушеннями в кінцівках в поєднанні з гіпертрофією периферичних нервів і іхтіозом.
3. Спадкова ПНП з нахилами до паралічів від здавлення являеться своєрідною рецидивуючою фокальною ПНП, успадкованою по аутосомно-домінантному типу. Починається у віці 20—30 років з рівною частотой у мужчин і жінок. Клінічна картина повязана з формуванням протікаючих безболючо гострих мононевропатій або множинних мононевропатій з руховими і чутливими порушеннями в кінцівках.
Частіше уражаються нерви в місцях їх найбільш типової компрессії після незначних травм і здавлення: •малостегновий (35%) •ліктьовий (20%)
•променевий (9%) Неврологічні симптоми зберігаються від декількох днів до декількох тижнів.З протіканням хвороби регресс сповільнюється з формуванням аміотрофій кінцівок.
При ЭНМГ виявляеться демієлінізучий характер ураження.
У більшості хворих прогноз благоприємний. Загальна характеристикаелектронейроміографічних показників при різних формах поліневропатій. (тема наступної публікації)